Molecular docking
Molecular docking is a technical method to simulate the interaction between ligand small molecules and receptor biological macromolecules based on the "lock key principle" of ligand receptor interaction. The interaction between ligand and receptor is a process of molecular recognition, mainly including electrostatic interaction, hydrogen bonding interaction, hydrophobic interaction, van der Waals interaction, etc. Through calculation, the combination mode and affinity between the two can be predicted. Yanjin Bio provides professional molecular docking services and a complete set of virtual drug screening services, which can help customers discover the ligand protein interaction mode, explain biological experiments, discover new active compounds and provide guidance for compound optimization. It covers protein preparation, active site discovery, protein flexible conformation exploration, ligand conformation database preparation, docking result analysis and evaluation, small molecule compound library preparation, pharmacophore modeling and screening, manual experience screening, etc.
Molecular docking applications:
1. To explore the specific mode of action and binding configuration of small and large drug receptors;
2. Screening the lead drugs that can bind to the target;
3. Explain the reason why drug molecules produce activity;
4. To guide rational optimization of drug molecular structure.
Molecular docking is one of the important methods of molecular simulation. Its essence is the recognition process between two or more molecules, involving the spatial matching and energy scoring between molecules. According to the energy ranking, the preliminary optimal structure and binding mode between molecules are finally obtained.

The docking software between "protein ligand" is complex, and Yanjin biological analysis software mainly includes Autodock, Vina, Dock, etc.
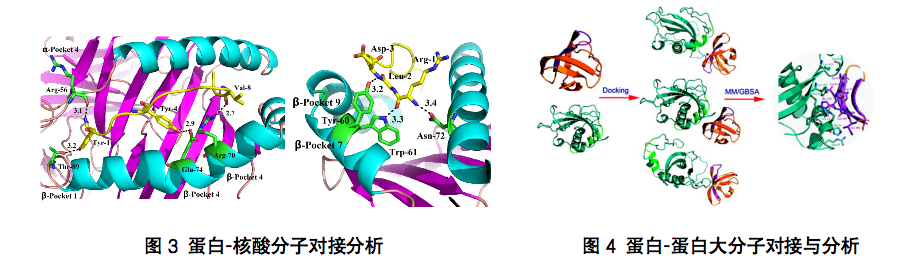
Molecular docking technology has a low threshold but is difficult to master, and it is easy to obtain false positive results. Accurate molecular docking results can not be separated from the knowledge and technology reserves and long-term first-line experience accumulation. Choose Yanjin Biology to let professional people do professional things. As long as you provide relevant biological information, we can help you find a reasonable combination mode and result analysis.
With the development of X-ray diffraction and NMR technology, a large number of three-dimensional crystal structures of target proteins have been resolved, which can not be directly used for molecular docking. In fact, in the process of protein parsing, there are often various errors, such as the loss of atoms, the incompatibility between the secondary sequence and the three-dimensional structure of the protein, which will affect the accuracy of docking, especially when these errors appear in the binding pocket of the ligand. Therefore, these errors must be corrected before docking. Both X-Ray and NMR can only determine the position of heavy atoms without the position information of hydrogen atoms. Before docking, hydrogenation protonation is required to mark the local electrical properties, so that it can be used for docking. After preparing the protein structure, it is necessary to find the active sites for drug molecule binding. The topology of the protein surface is very complex and diverse, and the physical and chemical properties are also extremely diverse. Which sites are the binding sites of small molecules of drugs, and can inhibit or activate the activity of the protein? In fact, there must be some corresponding research and annotation on the biological function of the target protein. In most cases, proteins also perform their biological functions by binding to natural ligands (macromolecules or small molecules). The binding sites of these natural ligands are likely to be the binding sites of their inhibitors or agonists. In the absence of these corresponding biological annotations, we can also use computational analysis to investigate the protein surface from multiple perspectives, such as topology, physical and chemical properties, to find appropriate binding sites, and combine with experimental information to finally determine the active sites.
As we all know, in the process of protein ligand interaction, there is an induced fit effect, and its conformation will change accordingly during the binding process. An accurate docking must take into account the flexibility of the receptor and ligand. Although many of the current software tools claim to be able to perform flexible docking of receptors, there are relatively large limitations in the methods, which may only be used to optimize the conformation of side chains through force field optimization and other methods. Yanjin Biology can investigate several different conformations that may exist in the protein by means of computer simulation. These conformations can be used as the starting point for docking to consider the flexibility of the protein more. The other flexibility is the flexibility of the ligand. Although the software will automatically consider the flexibility of the ligand during the docking process, such as rotating some rotatable keys. However, the generation of this conformation is also relatively limited, for example, the conformation of saturated rings cannot be fully considered. We can use conformational search, saturated ring conformational search and other methods to traverse the dominant conformations of ligands as much as possible to serve as the docking conformation library, so as to improve the accuracy! After docking, it is generally sorted by combining the score of free energy. Each ligand may have a variety of binding conformations. We select the most likely binding mode through comprehensive evaluation methods, such as the scoring of binding free energy, molecular stress energy, etc., and combine artificial judgment to find a truly reasonable binding mode.
- TEL:027-87288106
- Phone:15337192895/18963979811
- Email:[email protected]
- Address:Yuechuang center, Huazhong Agricultural University, Hongshan District, Wuhan
